Редкий синдром марфана: признаки, способы лечения и наследование у детей
Содержание:
Болезни радужной оболочки
Синдрому Марфана нередко сопутствуют изменения в радужной оболочке — это связано с повышенной растяжимостью тканей. Могут возникнуть колобомы, гипоплазия или атрофия радужки с нарушением ее диафрагмальной функции. Колобома — это дефект, проявляющийся в отсутствии части глазной оболочки. Обычно имеет грушевидную форму и располагается в нижней части радужки. К наиболее ранним проявлениям у людей с синдромом Марфана относят гипоплазию стромы радужки, особенно ее пигментной зрачковой каймы. Слабость дилататора (мышцы-расширителя) у больных не позволяет достичь полного расширения зрачка даже с помощью мидриатиков.
К какому врачу обратиться
При подозрении на наличие синдрома Марфана следует обратиться к врачу-генетику. После проведения диагностики к наблюдению и лечению пациента подключаются и другие специалисты: кардиолог, ортопед, офтальмолог и терапевт. При необходимости проведения корригирующих операций на сердце и сосудах больного направляют к кардиохирургу.
Синдром Марфана является редким генетическим заболеванием, которое сопровождается неправильным развитием соединительной ткани. Впоследствии у больного возникают поражения скелетных структур, сердца и сосудов, органов зрения, нервной и других систем организма. Постоянное врачебное наблюдение за такими пациентами, регулярное диагностическое обследование и своевременное лечение возникающих патологий позволяют существенно улучшать качество жизни таких людей и предупреждают развитие жизнеугрожающих осложнений.
Медицинская анимация о симптомах синдрома Марфана (англ. яз.):
О синдроме Марфана в программе «Жить здорово!» с Еленой Малышевой (см. с 30:20 мин.):
https://youtube.com/watch?v=OANPwgRoI48
Причины возникновения
Это аутосомно-доминантное состояние, генетическое заболевание, передается от родителей к ребенку через гены.
Вызван мутациями в гене FBN1. Мутации FBN1 связаны с широким континуумом физических функций, от изолированных особенностей до тяжелой и быстро прогрессирующей формы у новорожденных.
Особенности расстройства чаще всего обнаруживаются в сердце, кровеносных сосудах, костях, суставах и глазах. Некоторые особенности – например, расширение аорты (расширение основного кровеносного сосуда, которое переносит кровь от сердца к остальной части тела) – может быть опасным для жизни. Также могут быть затронуты легкие, кожа, нервная система. Патология не влияет на интеллект.
Распространенность, частота встречаемости составляет 1 из 5 000 человек, включая мужчин и женщин всех рас и этнических групп. Около 3 из 4 наследуют его, то есть получают генетическую мутацию от родителя, у которого она есть. Тип наследования при котором заболевший первый в семье- называется спонтанной мутацией. Существует 50-процентный шанс, рождения ребенка с генетической мутации от пораженных родителей.
Люди с синдромом Марфана рождаются вместе с ним, но особенности расстройства не всегда присутствуют сразу. У некоторых людей есть много особенностей при рождении, включая серьезные состояния, такие как расширение аорты. У других меньше симптомов, например, молодые не имеют признаков, пока не станут взрослыми. Некоторые особенности, особенно те, которые влияют на сердце и кровеносные сосуды, кости или суставы, со временем ухудшаются.
Это делает очень важным получение точной ранней диагностики и лечения. Без этого человек подвергается риску от потенциально опасных для жизни осложнений. Чем раньше началось лечение, тем лучше результаты.
Почти половина людей, страдающих синдромом Марфана, этого не знают.
https://youtube.com/watch?v=OANPwgRoI48
Что такое синдром Марфана
Синдром Марфана – совокупность симптомов и признаков, составляющих клиническую картину серьезного генетического наследственного заболевания, которое поражает связочные ткани
Точнее, синдром Марфана является следствием мутации гена FBN1, расположенного на хромосоме 15, которая кодирует фибриллин-1, гликопротеин (белок, который содержит полимер, простые сахара или олигосахарид), они имеют важное значение для эластичности волокон соединительной ткани
Таким образом, синдром Марфана влияет на множество важных органов и тканей: сердце, легкие, скелет, глаза и т.д.
Заболевание передается, как аутосомно-доминантный признак. Большинство больных наследует ген от одного родителей. Замечено, что эти случайные мутации имеют наиболее высокую вероятность проявления у пожилых родителей.
Взгляд на соединительную тканьСоединительная ткань выполняет функцию поддержки, объединения и питания тканей организма, которые образуют различные органы. С точки зрения гистологии (наука, которая изучает состав тканей) соединительная ткань может быть различных типов, в зависимости от характеристик ткани и/или органа, которые она образует, но всегда характеризуется наличием клеток, разделенных между собой, но рассеянных в межклеточном веществе как внеклеточный матрикс. Внеклеточный матрикс состоит из белков, которые образуют волокнистую часть фиброзной ткани и водный раствор белка. Волокнистая ткань может состоять из трех различных типов волокон: коллагеновых, сетчатых и эластичных. Сетчатые и коллагеновые волокна имеют идентичный химический состав, но отличается структурной организацией. Эластические волокна состоят из двух отдельных белковых цепей в фибриллина и эластина. Основными видами соединительной ткани являются:
|
Механизм, с помощью которого развивается синдром Марфана
Мы сказали, что Синдром Марфана является следствием мутации гена FBN1, который кодирует белок фибриллин-1. Он необходим для образования эластических волокон, которые составляют структуру соединительной ткани, а также содержат некоторые факторы роста, такие как TGF-beta.
Мутация FBN1 определяет снижение на физиологическом уровне фибриллина 1 и, следовательно, невозможность накопления TGF-beta и ухудшение волокон эластина, то есть возникновение синдрома Марфана.
Симптомы
Ведущие симптомы наблюдаются со стороны опорно-двигательного аппарата, сердца и глаз. Однако другие органы также страдают от дефектов соединительной ткани. В общем, для синдрома Марфана характерно следующее:
• Деформации грудной клетки со сколиозом, ограниченной дыхательной подвижностью легких.
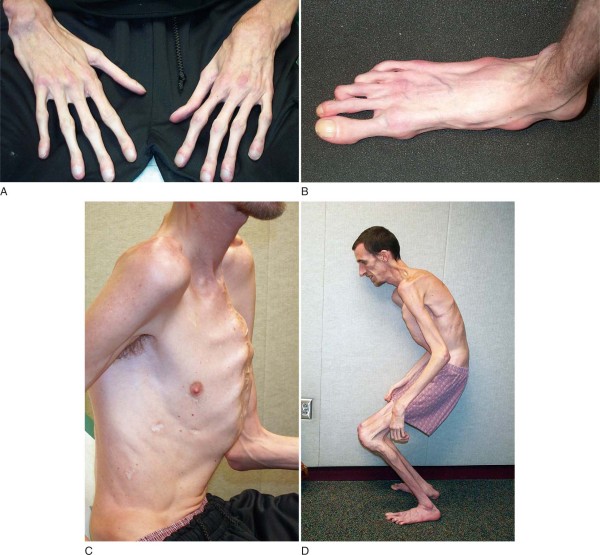
• Длинные конечности, непропорциональные туловищу, с широким охватом обеих рук, длинными и тонкими пальцами, узкими ладонями. Исключительная подвижность суставов. Ноги также удлиненные и плоские.
• Лицевой дисморфизм с удлинением головы, лица и носа. Ушные раковины деформированы и приближены к голове. Небо высокое, иногда отмечаются дефекты его закрытия или так называемой волчьей пасти. Глазные яблоки иногда опущены.
• Неправильное размещение линзы хрусталика, дефект роговицы, слабость цилиарных мышц (расположенных в радужной оболочке), неправильное развитие радужной оболочки, увеличение глазного яблока – факторы, приводящие к близорукости и повышенному риску отслоения сетчатки и потери зрения. Из-за слабости соединительной ткани склера у детей тонкая и голубоватая из-за прозрачного нижнего слоя глазного яблока.
Расширение корня аорты с возможной недостаточностью аортального клапана и возвращением крови в левый желудочек во время диастолы. Существует риск расслоения аорты в ее стенке – опасное для жизни состояние. Другие крупные сосуды также могут быть вовлечены в патологический процесс.
Выпадение митрального клапана является ведущим дефектом клапана. Другие клапаны также могут быть затронуты – либо сужены, либо неэффективно закрыты. Эти нарушения сердечной деятельности приводят к сердечной недостаточности с полной ее клинической картиной.
Пациенты жалуются на гипергидроз (повышенное потоотделение), и на их коже наблюдается характерный мраморный рисунок кожи. Пальцы часто голубоватые.
Психическое состояние детей с синдромом Марфана остается нормальным, а их интеллектуальное и эмоциональное развитие, несмотря на тяжелые заболевания, не отличается от таковых у их сверстников. Заболевание чаще всего выявляется сразу после рождения или до первого года жизни ребенка.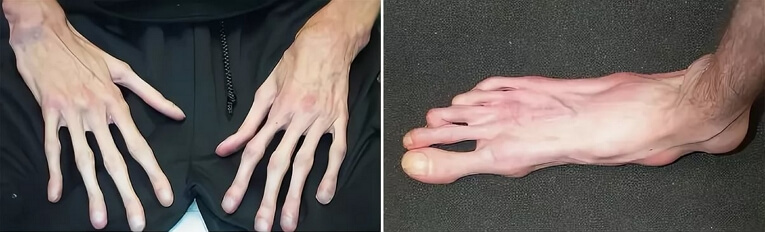
Методика лечения
Лечение направлено на профилактику прогрессирования и развития осложнений заболевания
Своевременная диагностика и лечение синдрома Марфана очень важна. Чем раньше начать лечение, тем более эффективным оно будет. Лечебные процедуры касаются всех 3 направлений развития осложнений: коррекция зрения, укрепление сердечной мышцы, нормализация формирования и развития костной ткани. Если больному ставится диагноз «синдром Марфана», его наблюдают сразу несколько врачей: терапевт, кардиолог, ортопед, кардиохирург, офтальмолог, генетик.
В первую очередь необходимо остановить развитие негативных последствий заболевания, нормализовать работу сердечно-сосудистой системы. В комплексное лечение обычно входят:
- Бета-адреноблокаторы. Это препараты, которые блокируют бета-адренорецепторы, что позволяет улучшить кровоснабжение миокарда, расширить просвет сосудов. Сердечная мышца активнее насыщается кислородом. При синдроме Марфана прием бета-адреноблокаторов начинается с раннего детства.
- Диета. Для укрепления сердечной мышцы и правильного роста необходимо употреблять большое количество продуктов, богатых магнием (зелень, свежие овощи).
- Аскорбиновая кислота. Для разжижения крови и поддержания тонуса сосудов пациенты с синдромом Марфана постоянно принимают аскорбиновую кислоту.
- Хлорид карнитина. Это препарат, который нормализует метаболизм и активизирует липидный обмен, назначается при различных нарушениях мозгового кровообращения.
- Хирургическое лечение аневризмы. Чаще всего больные с синдромом Марфана умирают в возрасте 30-40 лет по причине разрыва аневризмы. Чтобы продлить жизнь пациенту, рекомендуется операция по протезированию расширенной аорты. По статистике это позволяет продлить жизнь на 5-10 лет.
- Хирургическая коррекция зрения. Часто при синдроме Марфана страдает хрусталик. Нередко рекомендуется его удаление и протезирование.
Восстановить зрения получается далеко не всегда, но удается остановить развитие слепоты. Пациенту подбираются контактные линзы и очки для постоянного ношения. Также может назначаться операция по исправлению дефектов костей и суставов.
Последствия и профилактика
Синдром опасен своими осложнениями!
Синдром Марфана сопровождается множеством осложнений, приводящих к нарушению работы внутренних органов. Более 90% больных не доживают до 50 лет. Риск внезапной смерти при синдроме довольно высок. Это связано в первую очередь с расслоением аорты, разрывом аневризмы и прочими сердечными патологиями.
Из-за высокого роста и длинных конечностей многие люди с синдромом Марфана стремятся стать профессиональными спортсменами. Вероятность остановки сердца при высоких физических нагрузках особенно велика.
Среди последствий синдрома Марфана часто встречаются:
- Отслойка сетчатки. Сетчатка выстилает глазное яблоко изнутри. Ее отслойка является серьезным заболеванием, которое может привести к полной слепоте. Сначала отслойка сетчатки проявляется лишь в небольших мушках перед глазами. Затем появляется черная пелена, зрение начинает быстро ухудшаться. Единственным методом лечения является операция.
- Катаракта и глаукома. Очень часто у больных с синдромом Марфана наблюдаются сразу обе глазные патологии: и катаракта, и глаукома. При глаукоме повышается внутриглазное давление, что приводит к необратимым изменениям в нервном волокне. Катаракта сопровождается помутнением хрусталика глаза. В большинстве случаев лечение только хирургическое.
- Спонтанный пневмоторакс. Из-за деформации грудной клетки возникают проблемы с работой легких. Довольно часто искривленная грудная клетка не дает использовать весь объем легких. Это может привести к нарушению целостности плевры.
- Расслоение аорты. Это наиболее частое и смертоносное последствие синдрома Марфана, сопровождающееся поступлением крови между слоями аорты и ее разрывом.
О профилактике в данном случае говорить трудно, поскольку это заболевание связано с мутацией определенного гена. Предсказать или предотвратить развитие заболевания во внутриутробном периоде практически невозможно. Есть вероятность, что шанс возникновения заболевания снизится, если до беременности пройти обследование и посетить генетика.
Если заболевание уже есть, то профилактикой можно считать лишь регулярное посещение врача и умеренную физическую активность.
Симптоматика
При этом недуге формируются сочетанные изменения в опорно-двигательном аппарате, нервной и ССС, а также в органе зрения. Еще характерными признаками считаются разнообразные проявления, различные сроки формирования первых симптомов недуга и прогрессирующее течение в хронической форме.
Пациенты с данным патологическим состоянием характеризуются высоким ростом, относительно не большим телом с длинными не пропорциональными тонкими конечностями и паучьими пальцами. Они имеют худощавое телосложение со слабовыраженной подкожной клетчаткой и мышечный гипотонус, узкий и вытянутый скелет лица, изменение прикуса и высокое аркообразное небо. Средней рост у мужской половины при рождении составляет 53см, у женской 52,5см. А полный рост у мальчиков – 191см, у девочек- 175см.
При этом недуге происходит изменение функций подвижных соединений организма (повышенная гибкость, хруст в суставах и другое), деформируется грудина (например, куриная грудь или напоминает воронку), изменения позвоночного столба (кифоз, сколиоз и другое), а также появляется плоская стопа и протрузия вертлужной впадины.
Симптомы синдрома Марфана со стороны сердечно-сосудистой системы являются доминирующими и часто определяют исход течения заболевания. Проявляется изменения структуры сосудистых стенок эластичного вида (чаще аорты, крупные артерии легких, пороки развития клапанов и перегородок сердца). Дефекты аорты наблюдаются в виде расширения восходящего отдела и кольца клапана, а также в виде выпячивания стенки.
Симптомы синдрома Марфана со стороны митрального клапана заключаются в уплотнении створок, удлинение и разрыве хорд, обызвествление фиброзного кольца. В период беременности у плода возможно формирование врожденных пороков сердца (сегментарное сужение просвета аорты, сужение легочной артерии, дефект межжелудочковой и межпредсердной перегородки), а также воспалительный процесс внутренней оболочки сердца.
Наиболее неблагоприятная форма этого недуга является неонатальная, которая уже при рождении малыша приводит к быстрому развитию сердечно-сосудистой недостаточности и к смертельному исходу на 1 году жизни.
Также характерным признаком являются патологические состояния глаза: ненормальную рефракцию, смещение хрусталика из стекловидной ямки, изменения роговицы, недоразвитие радужки и цилиарной мышцы, страбизм, изменение просвета сосудов сетчатки. Смещение хрусталика при этой патологии происходит с двух сторон, чаще в возрасте до 4 лет и носит прогрессирующий характер, значительно ухудшая функцию зрения.
На ряду с этими нарушениями наблюдаются изменения и в иных системах и органах организма:
- Нервная – эктазия твердой мозговой оболочки;
- Бронхолегочной – скопление воздуха или газов в плевральной полости не связанное с травмой, чрезмерное скопление воздуха в легких, ОДН);
- Кожный покров – атрофические рубцы;
- Повторяющиеся грыжи в области паха и бедра, вывихи и надрывы связочного аппарата;
- Нарушение нормального положения почки, смещение мочевого пузыря и матки вниз, варикоз.
Также при этой патологии наблюдается повышенный выброс адреналина, что может привести больного в состояние постоянной возбудимости, двигательной активности, а в единичных случаях к неординарным способностям и умственной одаренности.
Симптоматика
Синдром Марфана проявляется разнообразными клиническими признаками, что связано с присутствием соединительнотканных волокон в различных структурах организма. У больных появляются признаки поражения костей и суставов, зрительного анализатора, сердца, сосудов, нервов, легких, кожного покрова.
Симптомы дисфункциональных расстройств внутренних органов возникают на фоне сильной усталости и быстрой утомляемости после незначительных физических нагрузок, миалгии, вялости, мышечной гипотонии, приступообразной цефалгии. По мере роста и развития организма человека черты заболевания становятся более выраженными.
Скелет
Клинические признаки синдрома Марфана, обусловленные поражением костей, мышц и связок:
- короткое туловище и длинные ноги,
- молоткообразные пальцы стопы,
- паучьи пальцы кисти,
- худощавое телосложение,
- гипотонус мышц,
- удлиненное и узковатое лицо,
- глубокая посадка глаз,
- отсутствие зазоров между зубами,
- большой нос,
- микрогнатия,
- недоразвитые скулы,
- большие и низко расположенные уши,
- «готическое» небо,
- прогения,
- гиперподвижность суставов,
- деформированная грудная клетка в виде воронки или киля птицы,
- искривление позвоночника,
- смещение вышележащего позвонка относительно нижележащего,
- уплощение сводов стопы,
- продавливание вертлужной впадины.
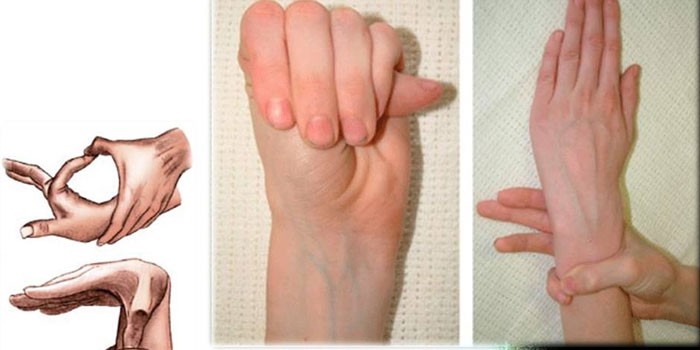
Сердце и сосуды
Поражение сердечной мышцы при синдроме Марфана определяет его исход. Обычно возникают дефекты в структуре аорты, легочного ствола, развиваются пороки развития клапанов сердца:
- дилатация предсердий и желудочков,
- мешкообразное расширение ограниченного участка аортальной стенки,
- пролабирование двустворчатого клапана,
- миксоматоз сердца,
- кардиомиопатия,
- разрыв хорд митрального клапана,
- кальциноз аортального клапана,
- различные виды аритмии – мерцательная, экстрасистолия,
- воспаление внутренней оболочки сердца – эндокарда,
- кардиалгия с иррадиацией в спину, ключицу, руку, плечо,
- похолодание конечностей,
- затрудненное дыхание,
- сердечные шумы,
- на ЭКГ — признаки ИБС.
Глаза
У лиц с синдромом Марфана развивается патология зрения:
- миопия,
- смещение хрусталика в сторону,
- уплощение роговой оболочки глаза и ее утолщение,
- недоразвитие радужки и ресничной мышцы,
- страбизм,
- катаракта,
- асимметричность зрачков,
- глаукома,
- астигматизм,
- дальнозоркость,
- колобома глаза,
- спазмирование сосудов сетчатки и высокий риск ее отслойки.
Прочие органы
- ишемические и геморрагические инсульты,
- субарахноидальное кровотечение,
- выпячивание оболочек спинного мозга.
Ослабление и растяжение дурального мешка проявляется болью в пояснице, ногах, тазу и другими неврологическими признаками, цефалгией.
- скопление воздуха между висцеральным и париетальным листками плевры,
- эмфизема легких,
- удлинение и перерастяжение альвеол,
- ночное апноэ,
- рак легких.
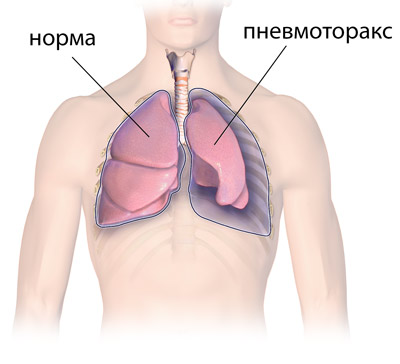
Поражение кожи, мягких тканей, внутренних органов:
- смещение почки в каудальном направлении,
- цистоцеле,
- пролапс матки,
- кисты и новообразования в печени и почках,
- варикоз,
- стрии на коже,
- грыжи.
Больные с синдромом Марфана легко возбудимы, гиперактивны, чрезмерно эмоциональны, часто плаксивы и разражительны. Они имеют неординарные способности и высокий уровень интеллекта.
«Готическое» небо и микрогнатия приводят к возникновению речевых нарушений. При поражении скелета и суставов появляются артралгии и миалгии, развивается ранний остеоартрит.
Синдром Марфана проявляется по-разному у лиц, обращающихся за медицинской помощью. У одних выявляются ярко выраженные признаки патологии, у других симптоматика бывает стертой. Прогрессирование синдрома происходит по мере взросления человека. Клинические проявления определяются локализацией и интенсивностью поражения органов и систем.
Методика коррекции аномалии
В детском лечении принимает участие множество препаратов, влияющих на сердечно-сосудистую систему, стимуляторы ЦНС, энерготропные препараты и антиоксиданты. Дополнительно назначают бета-адреноблокаторы: Обзидан, Атенолол, – которые требуется употреблять на протяжении года.
Среди эрготропных и антиоксидантных препаратов, которые назначаются для коррекции аномалии, можно отметить:
- Рибоксин;
- Витамины В1 В2;
- Аскорбиновая кислота ;
- Токоферол (вит Е);
- Элькар;
- Димефосфон;
- Коэнзим Q10;
- Лимонтар;
- Пирацетам.
Дозировка препаратов врачом определяет индивидуально, учитываются особенности развития и характер выраженности патологий. То же самое касается длительности лечения, но в большинстве случаев для достижения оптимальных результатов требуется корректирующая терапия на протяжении нескольких лет.
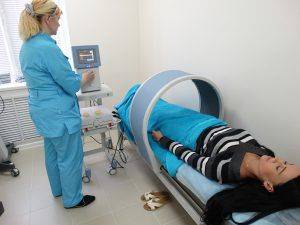 Магнитотерапия
Магнитотерапия
Кроме медикаментозной терапии, врачи назначают дополнительные курсы лечения с использованием магнитотерапии на суставы, электросна, лечебной физкультуры, которая воздействует преимущественно на опорно-двигательный аппарат. Пациенты отправляются на санаторно-курортное лечение для больных с нарушениями функций костей и суставов. Иногда проводится оперативное лечение и осуществляется регулярная санация очагов инфекции в полости рта и зубов.
Для того чтобы отмечалась положительная динамика в процессе лечения ребенка с синдромом Марфана, требуется соблюдать следующие рекомендации:
- Разрешать ребенку заниматься физкультурой, но по специальной ослабленной программе. Известно, что люди с синдромом Марфана отличаются высоким ростом и удлиненными конечностями. Ошибочно полагая, что их организм вынесет большие нагрузки, они стремятся стать звездами в спорте, от чего и умирают в молодом возрасте. Несмотря на характерную внешность, которая идеальна для баскетболистов и бегунов, сердечно-сосудистая система этих людей очень ослаблена, и чаще всего больные люди умирают от разрыва аорты.
- Ношение тяжестей, прогулки по горной местности, сельскохозяйственные работы, участие в марафонах и спортивных состязаниях находятся под строгим запретом.
- Запрещается длительное контактирование пациентов с химическими веществами и нахождение в помещениях, где пахнет лаками, красками и бытовой химией.
- При выборе места жительства лучше избегать жаркой местности и зон с повышенным уровнем радиации.
В качестве дополнительной терапии применяется специальная диета, которая включает повышенное количество магния. Доктор в индивидуальном режиме, обязательно с учетом предпочтений пациента, разработает оптимальную схему питания, которая позволит компенсировать недостаток магния в организме. Дети и взрослые с синдромом Марфана должны постоянно быть под присмотром врачей и проходить плановое обследование минимум 2-3 раза в год.
К примеру, недавно было уставлено, что главную причину смертности среди Марфанов, заключающуюся в разрыве аорты, можно предупредить препаратом для лечения давления – Лозартаном. На данный момент ведутся исследования, способен ли препарат устранить и риск аневризмы аорты у пациентов.
Классификация
Согласно МКБ-10, синдром Марфана входит в класс врожденных дефектов развития и хромосомных патологий, тут патологию можно найти под шифром Q87.4.
Детальной клинической классификации недуга на сегодняшний день не существует, но выделяют несколько форм болезни, в зависимости от тех или иных критериев.
В зависимости от выраженности симптомов:
- стертая форма – признаки патологии мало выражены и могут оставаться незамеченными на протяжении всей жизни, как правило, изменения касаются не более 2 систем органов;
- клинически выраженная форма – симптомы патологии хорошо заметны и встречаются более чем в 2 системах органов.
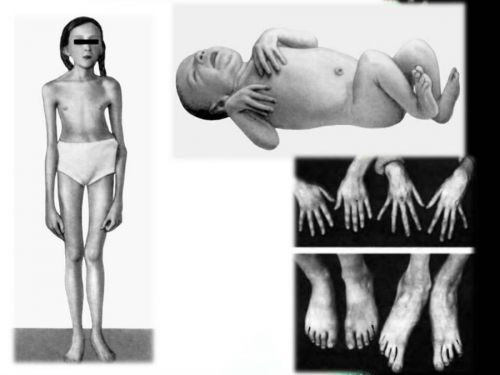 Внешний вид ребенка, больного синдромом Марфана
Внешний вид ребенка, больного синдромом Марфана
В зависимости от генетического фактора:
- семейная форма диагностируется в случаях, когда болезнь передается по наследству;
- спорадическая форма определяется тогда, когда патология обусловлена новой спонтанной мутацией у индивида и при этом не встречается у его родственников.