Синдром паучьих пальцев
Содержание:
Лечение
Поскольку все виды заболевания, сопровождающиеся арахнодактилией, являются генетическими, лечение назначается симптоматическое.
При синдроме Марфана назначаются:
- витамин С в больших дозах;
- анаболические стероиды (рибоксин), ускоряющие синтез белка в клетках;
- энерготропные препараты (витамины В1 и В2, Коэнзим Q10 и др.), воздействующие на ключевые этапы клеточного энергообмена;
- антиоксидантные препараты (токоферол и др.), влияющие на биосинтез и метаболизм;
- ноотропные препараты, стимулирующие деятельность ЦНС (пирацетам).
Также назначают:
- элькар, повышающий работоспособность и выносливость;
- димефосфон, нормализующий кровоток и усиливающий энергетические процессы в мозге;
- лимонтар, регулирующий обменные процессы;
- атенолол или обзидан (бета-адреноблокаторы), снижающие частоту и силу сердечных сокращений и препятствующие расширению аорты.
При гомоцистинурии:
- для снижения риска образования тромбов в небольших дозах назначаются препараты, препятствующие свертыванию крови;
- назначается фолиевая кислота, участвующая в синтезе аминокислот;
- назначается бетаин, активирующий метаболическое метилирование в печени.
При В6-резистентной форме заболевания назначается низкобелковая диета и лишенные метионина аминокислотные смеси (XMET Maxamaid и др.).
При В6-зависимой форме назначают большие дозы пиридоксина гидрохлорида, восполняя таким образом дефицит витамина В6.
В зависимости от сопутствующих симптомов больным назначают ноотропы, способствующие усваиванию фолиевой кислоты и витамина В гепатопротекторы, препараты железа и кальция.
Арахнодактилия при любой этиологии заболевания является показанием к ЛФК и нуждаются в курсах массажа. По показаниям назначаются лазерная акупунктура и рефлексотерапия.
При выявлении патологии, которая существенно ухудшает состояние пациента или угрожает его жизни, проводят необходимую операцию. Это может быть протезирование сердечных клапанов или аорты (при диаметре больше 5 см), хирургическая коррекция формы грудной клетки, эндопротезирование тазобедренных суставов и т.д.
Коррекция зрения осуществляется методом подбора контактных линз или очков, а при необходимости методом лазерного или хирургического лечения.
Что такое «синдром Марфана»?
Синдромом Марфана называют редкое генетическое заболевание, которое наследуется по аутосомно-доминантному типу. Оно обусловлено структурными дефектами соединительной ткани. Всё это приводит к поражению опорно-двигательной, сердечно-сосудистой и зрительной системы организма. Коллаген и патология встречаются не очень часто. Согласно статистике, один случай возникает на 10000-20000 человек. Патология может появиться у представителей любого пола. Характерная расовая определенность также отсутствует.
Впервые заболевание описал французский педиатр Антуан Марфан. Действие было осуществлено в 1886 году. Именно он рассмотрел клинический случай девочки, которая имела паучьи пальцы и длинные тонкие ноги. Вышеуказанные аномалии скелета прогрессировали. Процесс осуществлялся очень быстро. В результате заболеванию было присвоено имя исследователя. Ген, который провоцирует развитие признаков синдрома, впервые был выявлен только в 1991 году. Действия совершил Франческо Рамирес.
Заболевание развивается из-за того, что в результате генетической мутации нарушается синтез фибриллина, а также происходит формирование одновременно нескольких фенотипических признаков. Изменения могут происходить под воздействием негативных эндогенных и экзогенных факторов. Белок фибриллин входит в состав многих структур организма. Если наблюдается его недостаток, соединительная ткань теряет свою прочность и эластичность. Всё это сказывается на состоянии сосудистых стенок и связочного суставного аппарата.
Клинические проявления синдрома весьма полиморфны. Люди с патологией могут иметь высокий рост, удлиненные и тонкие пальцы, очень длинные конечности, которые выглядят непропорционально по отношению к остальному телу. У таких пациентов арковидное, астеническое телосложение и глубоко посаженные глаза. Череп людей, страдающих болезнью Марфана, имеет удлиненную форму. Нередко присутствует деформация грудной клетки. Иными патологиями, которые провоцируются заболеванием, выступают эктопия хрусталика, близорукость, протрузия вертлужной впадины, плоскостопие, аневризма аорты. Дополнительно присутствует кифоз и сколиоз. Челюсть у таких пациентов маленькая.
Вышеуказанный фенотипический набор может быть практически не выражен. Подобное возможно при легких формах заболевания. Такие люди практически не отличаются от здоровых. В случае существенного развития патологии наблюдается сильное изменение внешнего вида человека. Заболевание прогрессирует крайне быстро. Такое количество вариантов обусловлено тем, что мутации в гене FBN1 и некоторых других генах (TGFBR-2 и др.) могут быть крайне разнообразны.
Интересный факт. Патология нередко встречается и у знаменитых людей. Известно, что патологий страдали известный скрипач Никколо Паганини. Знаменитый сказочник Ганс Христиан Андерсен, президент США Авраам Линкольн и композитор Сергей Рахманинов также страдали недугом. Аналогичная патология выявлена у вокалиста Ramones Джоуи Рамона, певца Троя Сивана и легендарного пловца Майкла Фелпса. Список можно продолжить. Предполагается, что неординарные способности таких людей могут быть вызваны характерным для заболеваний высоким выбросом адреналина. Он приводит к возникновению гиперактивности и в ряде ситуаций способен вызвать развитие талантов.
Согласно статистике, в 75% случаев заболевание является наследственным. Только в 15-25% случаев мутация появляется впервые. Специалисты выявили тот факт, что риск рождения ребёнка с подобной патологией повышается, если возраст отца в момент зачатия больше 35 лет.
Особенности заболевания
Болезнь Пертеса не такая уж редкость. На её долю приходится 18% от всех суставных патологий. Стоит отметить, что недуг также встречается у собак мелких пород.
https://youtube.com/watch?v=TEcy-pla_Ag
Причины возникновения
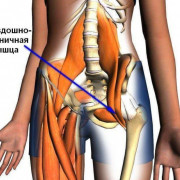
Истинные причины развития болезни до сих пор неизвестны. Но учёные выдвигают несколько предположений появления патологии. Так, основным определяющим фактором выступает миелодисплазия — недоразвитие в спинном мозге. Проходимость и количество кровеносных сосудов, обеспечивающих головку бедренной кости кровью, недостаточное.
Другие факторы, которые могут привести к началу болезни:
- Травмы тазобедренных суставов;
- Повышенные физические нагрузки;
- Анатомические особенности структуры тазобедренного аппарата;
- Нарушение обменных процессов (усвоение фосфора, кальция);
- Гормональные изменения в организме (переходный возраст);
- Инфекционное поражение суставов (например, после перенесённого синусита, ангины, отита).
Обычно начальные признаки появления недуга родители замечают через некоторое время после перенесённой простуды или травмы ребёнком. Именно это становится решающим фактором возникновения заболевания у детей с нарушенным кровообращением в области тазобедренного сустава. Стоит отметить, что наследственность также играет определённую роль в развитии болезни Пертеса, но далеко не главную.
Врачи и учёные заметили, что болеют в основном мальчики (особенно недоношенные или рождённые с малой массой тела). В свою очередь, у девочек патология протекает в более тяжёлой форме. Чаще болезнь обнаруживается в возрасте 3−5 лет, реже — 13−15 лет.
Стадии и симптомы
В течение болезни Пертеса выделяют 5 следующих друг за другом стадий:
- Остеонекроз. Именно в этот период начинает развиваться прекращение снабжение кровью головки бедренной кости и появляться очаговый некроз. Поражается до 10% всей костной массы головки бедра. Как правило, синдром Пертеса у детей протекает без симптомов. Очень редко появляются слабые нарушения в походке ребёнка и несильные боли в области бедра.
- Импрессионный перелом головки бедра. Под воздействием обычных нагрузок со временем разрушается область костной ткани, развивается перелом и образуется деформация бедренной головки кости. Площадь изменений может достигать 10−30%. Появляется болевой синдром и дискомфорт в области колена и бедра при движении, ребёнок при ходьбе прихрамывает.
- Фрагментация. Часть изменённой кости начинает распадаться на части. Площадь повреждения увеличивается до 30−50%. При этом боль сильно выражена и не проходит даже в спокойном состоянии. Нарушается двигательная активность в бедре, опухают мягкие ткани. Изменяется походка, хромота ярко выражена. У некоторых больных повышается температура, появляются симптомы интоксикации.
- Репарация. Над разрушительными изменениями постепенно преобладают восстановительные и регенерирующие процессы. Между некоторыми частями костной ткани начинает образовываться соединительная ткань, формируется новая костная ткань и кровеносные сосуды. Структура новой кости почти обычная, но её механическая прочность очень слабая. На этой стадии также начинает расти головка бедра. При отсутствии лечения это может стать причиной последующих изменений, так как головка способна перерасти в неправильную сферическую форму.
- Исход. На стадии исхода развиваются последствия заболевания (если они присутствуют). Возможно, и окончательное выздоровление без остаточных явлений. В этот период очень важна целостность хрящевой части роста. В случае некроза эта часть способна частично или полностью разрушаться. В итоге изменяется походка и нарушается опорная функция конечностей.
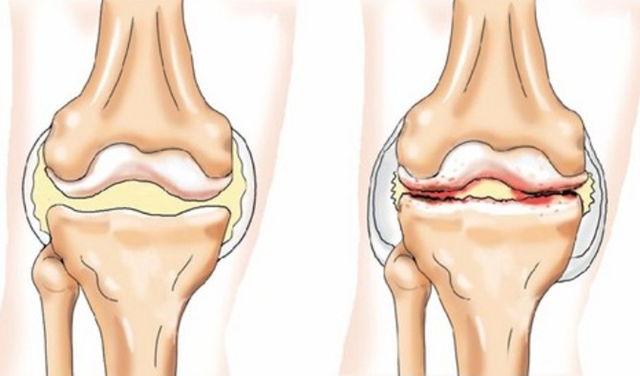
Если деформация головки бедра формируется на четвёртой стадии, то в этом случае развивается артроз тазобедренного сустава.
Арахнодактилия (синдром паучьих пальцев): причины и признаки патологии, диагностика и методы терапии, общие сведения о болезни и ее формы
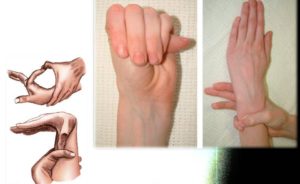
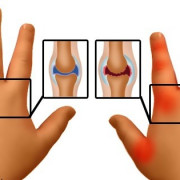
Паучьи пальцы (арахнодактилия) — это заболевание, при котором они становятся длинными и вытянутыми, приобретают характерную форму. В некоторых случаях они отличаются повышенной гибкостью и могут отклоняться полностью назад.
Это врожденная патология, которая может проявляться отдельно или в комплексе с другими признаками генетических заболеваний.
Сама по себе она не представляет опасности, но требует обязательной диагностики — синдром паучьих пальцев часто протекает совестно с более серьезными нарушениями работы внутренних органов.
Методы лечения
Поскольку арахнодактилия является генетическим заболеванием, полностью избавиться от ее симптомов не получится. Однако можно применять симптоматические методы для улучшения состояния пациента и профилактики осложнений. Препараты подбирают индивидуально, в зависимости от симптомов болезни и их тяжести.
При болезни Марфана можно назначать следующие средства:
- витамин С в повышенной дозировке;
- стероидные средства для улучшения процессов синтеза белка;
- лекарства, которые воздействуют на обмен энергии (витамины группы В, кофермент Q10);
- антиоксиданты (витамин Е);
- ноотропные средства для активизации деятельности головного мозга.
При гомоцистинурии можно применять лекарственные средства других групп:
- препараты для разжижения крови и предотвращения образования тромбов;
- фолиевая кислота;
- бетаин для стимуляции процессов метилирования в печени.
При этом заболевании также стоит учитывать его форму. Витамин В6-зависимая форма нуждается в постоянном введении этого вещества для стабилизации состояния здоровья. Если течение болезни не зависит от концентрации этого витамина, необходимо соблюдать диету с низким содержанием белка.
Вне зависимости от того, какое заболевание спровоцировало появление арахнодактилии, она нуждается в постоянной симптоматической терапии. Для стабилизации состояния больного полезно периодически проходить курсы массажа и лечебной физкультуры. Кроме того, по показаниям могут быть назначены физиопроцедуры — лазерная терапия и рефлексотерапия.
Оперативное вмешательство показано только в тех случаях, когда генетические аномалии существенно ухудшают качество жизни пациента. По показаниям могут быть назначены следующие операции:
- протезирование клапанов сердца или аорты;
- хирургическое изменение формы грудной клетки;
- восстановление тазобедренных суставов путем вживления эндопротезов.
Поскольку у многих пациентов с подобными диагнозами одновременно наблюдается ухудшение зрения, они вынуждены постоянно носить очки или контактные линзы. Также может быть назначена операция по восстановлению зрения или лечение лазером.
https://youtube.com/watch?v=5TUss-4Bbww
Существуют специальные генетические тесты, которые позволят определить вероятность рождения ребенка с арахнодактилией
Прогноз и профилактика
Прогноз при арахнодактилии зависит от основного заболевания, симптомом которого она является. Если своевременно выявить патологию и начать лечение еще в раннем детстве, можно избежать опасных осложнений. Особенно эффективна комплексная терапия при гомоцистинурии — у 70—80% детей с таким диагнозом можно добиться значительных улучшений благодаря приему специфических препаратов.
Все патологии, которые сопровождаются появлением паучьих пальцев у ребенка, представляют собой генетический сбой. В 75% случаев они связаны с семейным типом наследования, то есть гены этой болезни присутствуют у одного или у двух родителей. При планировании беременности стоит проконсультироваться у генетика, особенно если в анамнезе родственников есть подобные заболевания.
Паучьи пальцы — это один из симптомов многих заболеваний. Он может проявляться отдельно или в сочетании с другими патологиями. Эти нарушения представляют собой генетические мутации, поэтому не поддаются лечению. Поддержать состояние больного можно симптоматическими препаратами.
Арахнодактилия
Арахнодактилия (буквально «пальцы паука») – деформация пальцев кисти, наблюдаемая при некоторых наследственных патологиях. Как правило, арахнодактилии сопутствуют удлинение трубчатых костей (что приводит к деформациям скелета), патологические изменения сердечно-сосудистой системы и глазных яблок.
Главный внешний признак арахнодактилии – тонкие и длинные пальцы
Что такое синдром Марфана
Синдром Марфана – это наследственная патология, в результате прогрессирования которой происходит поражение соединительной ткани. В процесс также вовлекается скелетно-мышечная система и зрительный аппарат. Ткань внутренней среды (соединительная) выполняет массу важных функций, одной из которых является соединение определенных частей тела, и закладывание основы для их нормального роста и развития.
В случае прогрессирования синдрома Марфана, в структуре ткани появляются дефекты, которые не дают ей функционировать так, как необходимо.
- Причины
- Классификация
- Симптоматика Скелет
- Зрение
- Сердце и сосуды
- Нервная система
- Кожный покров
- Легкие
Диагностика
Лечение
- скелет;
- сердце;
- сосуды;
- зрительный аппарат;
- кожный покров;
- легкие;
- ЦНС.
Синдром Марфана в одинаковой мере поражает и мужчин, и женщин. Патологию диагностируют у людей всех этнических групп и рас. Распространенность – 1 случай на 10 тысяч человек. Риск рождения ребенка с данной болезнью повышается в несколько раз в том случае, если отец достигает возраста 35 лет. В 50% случаев ребенок рождается с патологией, если у одного из родителей диагностирован синдром Марфана. Тип наследования – аутосомно-доминантный.
Клиническая картина
Арахнодактилия затрагивает пальцы на руках, меняя не только форму, но и размер кисти. Пальцы становятся непропорционально тонкими, если их сравнивать с ладонью.
Они более длинные и изогнутые. В области межфаланговых суставов наблюдается типичная деформация. Все эти особенности придают руке типичный вид, который также получил название «паучьих пальцев». Развиваются данные признаки постепенно по мере прогрессирования основной патологии.
В ряде случаев помимо типичной формы пальцы также становятся более подвижными. То есть один или сразу несколько способны отклониться в тыльную сторону кисти практически на 180°. В большинстве случаев подобное патологическое удлинение протекает параллельно другим изменениям в скелете, органах и системах. Характер нарушений будет определять первичная патология, спровоцировавшая их.
Если говорить о синдроме Марфана, то арахнодактилия развивается параллельно иным изменениям в скелете. Конечности становятся тонкими, непропорциональными телу, длинными, что во многом достигается именно за счет негативного воздействия на все трубчатые кости. Также страдает и связочный аппарат, который существенно слабее, чем у здорового человека. Это приводит к гипермобильности суставов.
Мышцы атрофированы и под кожей практически незаметны, как и жировой слой. Череп кажется удлиненным, из-за чего размер головы становится больше нормы. Лоб высокий, лобные бугры имеют выраженный характер, затылок несколько уплощен. Нижняя челюсть кажется либо недоразвитой, либо наоборот – массивной. Тазовые кости являются более плоскими в сравнении со здоровым человеком, грудная клетка имеет килеобразную либо воронкообразную форму, позвоночный столб искривлен. Состояние сочетается с выраженным плоскостопием.
Важно! Помимо типичных изменений в скелете присутствуют при синдроме Марфана также и нарушения со стороны сосудов с сердцем, зрения
Патогенез синдрома Марфана
Более половины веса человека представлено соединительной тканью, из неё состоит наша главная опора — скелет, внешние покровы — кожа. Сосуды, кровь и лимфа тоже состоят из соединительной ткани.
К клеткам соединительной ткани относятся фибробласты и их разновидности (остеобласты, хондроциты, одонтобласты, кератобласты), макрофаги (гистиоциты) и тучные клетки (лаброциты).
Мезенхима — проводник конституциональных, генетических и эпигенетических составляющих жизни человека. Патология соединительной ткани детерминирует определенное патологическое действие на весь организм в целом, на его физиологию и его конституциональные особенности.
При болезни Марфана происходит замена нуклеотидов в гене, содержащем информацию о структуре пептида фибриллина-1. Этот белок относится к гликопротеидам, принимает участие в микрофибриллярном комплексе, он обеспечивает основу эластических фибрилл соединительной ткани.
Межклеточный матрикс позволяет соединительной ткани поддерживать постоянную структуру, в нём находится огромное количество факторов роста, которые обеспечивают постоянное обновление клеток.
В крупных сосудах, связочном аппарате содержится большое количество эластиновых фибрилл, поражение которых и даёт основные клинические проявления синдрома Марфана.
При синдроме Марфана значительно поражается трансформирующий фактор роста бета (TGF-β), нарушается связывание его неактивной формы, что приводит к повышению биоактивности данного фактора, с чем связано появление многих проявлений болезни.
Патология фибриллина приводит к патологии формирования волокон, что вызывает утерю прочности и эластичности кожи и других соединительнотканных структур.
Изменение структуры коллагеновых волокон приводит к нарушению первичного звена гемостаза у пациентов с синдромом Марфана.
Имеются данные о дефектах мембранных и цитоплазматических механизмов проведения сигнала непосредственно в самом тромбоците, приводящих к нарушениям агрегации (объединения). Показано наличие самостоятельного мембранного дефекта тромбоцитов, протекающего с нарушением реакций высвобождения и транспорта внутриклеточного кальция.
Эластические фибриллы имеют вполне определенные механизмы участия в системе гемостаза. В сосудах с низкой скоростью сдвига происходит адгезия («прилипание») тромбоцитов к эластину через фибронектин. Регистрируется снижение его уровня в крови у людей с синдромом Марфана. Фибронектин, в свою очередь, образуется в клетках эндотелия и участвует в последующих репаративных процессах, создавая основу для производства других компонентов соединительной ткани — фибробластов. Таким образом, совершенно неоспоримо участие сосудистой стенки в реакциях свертываемости крови, и неизбежен вывод о возможных патологиях протекания нормальных гемостатических процессов при изменении состояния её структурных компонентов и процессов сосудистой регуляции.
Отмечена роль гормонального дисбаланса в развитии и усугублении дефектов соединительнотканных структур.
Тромботические проявления детерминированы нарушением реологии (вязкости) крови в патологически извитых сосудах брахиоцефальной зоны.
Поражение желудочно-кишечного тракта детерминировано тем, что эта система богата коллагеном. Наблюдаются дискинезия билиарного тракта по гипомоторному типу, грыжи пищеводного отверстия диафрагмы, аномалии желчных путей, долихосигма, хронический гастродуоденит со стёртой клинической картиной, склонностью к торпидному течению.
Симптомы
Арахнодактилия при любом вызвавшем ее заболевании проявляется изменением длины и формы пальцев. Во многих случаях пальцы кисти и стопы не просто тонкие и длинные, но и искривленные, поэтому в сочетании с укороченными сухожилиями напоминают когтистую лапу. Такие изменения наблюдаются с момента рождения, но характерный вид конечности и тело в целом приобретают к 3 — 4 годам.
В большинстве случаев у новорожденных длина тела значительно превышает стандартные показатели.
В длину также увеличиваются предплечья и голени, поэтому конечности выглядят тонкими и непропорционально длинными. Расположенная высоко надколенная чашечка усугубляет нарушение пропорций тела, поэтому и талия оказывается расположенной выше нормы.
Наблюдаются также во многих случаях:
- воронкообразная или сдавленная по бокам по типу птичьей груди грудная клетка;
- сколиоз или кифосколиоз;
- долихоцефалический (длинный и узкий) череп и изменения его лицевой части (высокий лоб с ярко выраженными буграми, слабо выраженная или выпяченная вперед нижняя челюсть и др.);
- деформация пяточной кости;
- врожденный вывих бедра;
- плоскостопие;
- патологические наросты на костной ткани (остеофиты).
Возможно также наличие привычного вывиха (повторного смещения) плечевой кости, надколенной чашечки и радиоульнарного сустава.
Жировой слой выражен минимально.
При синдроме Марфана арахнодактилия сопровождается:
- подвывихом хрусталиков кверху и их дрожанием, голубоватостью склер и близорукостью, не сопровождающейся развитием глаукомы;
- патологиями сердца и крупных сосудов, проявляющихся в поражении сердечных клапанов, пороках сердца, аневризме аорты;
- выраженным отставанием массы тела от роста больного;
- выраженной избыточной подвижностью суставов;
- арковидным небом.
Интеллект сохранен, но возможно наличие анизокории, нистагма, асимметрии сухожильных рефлексов, пирамидных расстройств.
Возможно также наличие геморрагического синдрома (кровоточивость слизистых оболочек и кожные кровоизлияния). Может развиться спонтанный пневмоторакс (воздух проникает в плевральную полость из-за нарушения целостности поверхности легкого), который наблюдается у 5 % больных.
При гомоцистинурии арахнодактилия сопровождается:
- снижением интеллекта у 2/3 пациентов вследствие нарушений нервно-психической деятельности (снижение IQ до 32-72 единиц, низкая работоспособность, упрощенная речь, дислалия, ослабление мимики, сложности с переключением внимания и др.);
- судорожным синдромом;
- подвывихом хрусталиков книзу, прогрессирующими поражениями глаз (астигматизм, отслойка сетчатки и др.) и развитием вторичной глаукомы;
- поражением артерий среднего диаметра (почечных, мозговых, венечных);
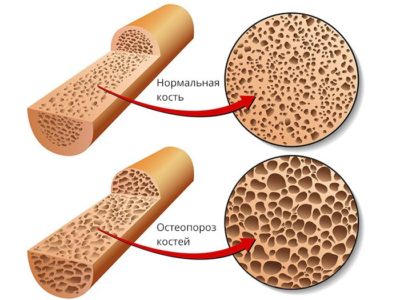
- остеопорозом;
- готическим небом;
- артериальным тромбозом (в половине случаев).
Плоскостопие, снижение массы тела и избыточная подвижность суставов выражены умеренно.
Для больных гомоцистинурией характерны светлые волосы, светлая кожа и голубой оттенок глаз.