Спинальная амиотрофия верднига-гоффмана: причины, симптомы, лечение
Содержание:
Классификация
Общепринятым считается разделение спинальных мышечных атрофий на детские и взрослые. Детские СМА классифицируются на ранние (дебютирующие в первые месяцы жизни), более поздние и ювенильные. Детские спинальные амиотрофии представлены:
- амиотрофией Верднига-Гоффманна;
- ювенильной формой Кугельберга-Веландера;
- хронической инфантильной СМА;
- синдромом Виалетто-ван Лэре (бульбоспинальная форма с глухотой);
- синдромом Фацио-Лонде.
Взрослые формы СМА манифестируют в возрасте от 16 до 60 лет и отличаются более доброкачественным клиническим течением. К СМА взрослого возраста относятся:
Выделяют также изолированные и сочетанные спинальные амиотрофии. Изолированные СМА характеризуются преобладанием поражения спинальных мотонейронов, которое во многих случаях является единственным проявлением заболевания. Сочетанные спинальные амиотрофии представляют собой редкие клинические формы, при которых симптомокомплекс амиотрофии комбинируется с другой неврологической или соматической патологией. Описаны сочетания СМА с врожденными пороками сердца, глухотой, олигофренией, понтоцеребеллярной гипоплазией, врожденными переломами.
Прогноз
Прогноз всецело зависит от клинического варианта СМА и возраста ее манифестации. Наиболее неблагоприятный прогноз имеют детские спинальные амиотрофии, при начале в младенческом возрасте они зачастую приводят к летальному исходу в течение первых 2-х лет жизни ребенка. Спинальные амиотрофии взрослого возраста отличаются способностью больных самостоятельно обслуживать себя в течение многих лет, а при медленном прогрессировании имеют благоприятный прогноз не только для жизни, но и для трудоспособности пациентов (при создании для них оптимальных условий труда).
Продолжительность жизни и прогноз были неутешительными до тех пор, пока на фармацевтический рынок не вышло инновационное лекарственное средство «Спиназа». Смерть многих новорожденных наступала сразу после появления на свет или на протяжении первого месяца жизни. При 1 типе заболевания нужна была постоянная респираторная поддержка. Максимальный срок жизни был около 1-2 лет.
Среди негативных последствий, которые часто диагностируются у детей и взрослых с диагнозом спинальная мышечная атрофия:
- бронхопневмония;
- аспирационная пневмония;
- острая дыхательная недостаточность.
Не нужно отчаиваться, если малышу поставили такой диагноз. Многие дети проживают 10 и более лет. Однако для достижения максимальных терапевтических результатов важен постоянный уход и круглосуточное наблюдение за ребенком, соблюдение назначений и рекомендаций лечащего врача.
Уход и лечение
Клиническое ведение больного с СМА варьируется в зависимости от тяжести и типа. Лечение отдельных пациентов с одним и тем же типом болезни может варьироваться. При наиболее тяжелых формах (типы 0/1) люди имеют наибольшую мышечную слабость, требующую немедленного вмешательства. При легких формах, которые проявляются во взрослом возрасте, больные могут продолжительное время не обращаться за помощью.
Респираторный уход
Поражение респираторной системы является наиболее частой причиной смерти больных с первым и вторым типом заболевания. Спинальная амиотрофия типа 3 может давать схожую симптоматику, но очень редко. Осложнения возникают из-за того, что прекращается иннервация межреберных мышц, являющихся важными участниками процесса дыхания. Диафрагма поражается меньше.
После ослабления нервной регуляции мышцы никогда полностью не восстанавливают функциональную способность, чтобы участвовать в акте дыхания, как прежде. Таким образом, дыхание представляет трудности, возникает риск нехватки кислорода, поверхностного дыхания и недостаточного очищения дыхательных путей. Эти проблемы чаще всего возникают во время сна, когда мышцы наиболее расслаблены. Слабость глотательной мускулатуры, совмещенная с невозможностью полноценно кашлять приводит к аспирации мелких частичек, а затем пневмонии.
Чтобы помочь в дыхании, часто используется неинвазивная вентиляция, трахеостомия может иногда выполняться в более тяжелых случаях; оба метода вентиляции продлевают выживаемость в равной степени.
Удаление мокроты из дыхательных путей производится вручную (с помощью специального оборудования), назначается физиотерапия.
Питание
Чем тяжелее течение болезни, тем больше вероятность возникновения проблем со здоровьем, связанных с питанием. Они включают трудности с кормлением, жеванием и глотанием, невозможностью закрытия рта. Люди с такими трудностями могут подвергаться повышенному риску переедания или недоедания.
Другие трудности, особенно у больных с тяжелыми формами заболевания включают желудочную непроходимость, желудочный рефлюкс, запоры, рвоту и вздутие живота. В связи с этим может возникнуть необходимость иметь трубку для кормления или гастростому.
Кроме того, метаболические нарушения, возникающие в результате спинальной мышечной атрофии, ухудшают β-окисление жирных кислот в мышцах и могут привести к органической ацидемии и последующему разрушению мышц, особенно при голодании.
Врачи рекомендуют больным, у которых спинальные амиотрофии, с прогрессирующей симптоматикой употреблять продукты с невысоким содержанием жиров, предпочесть более жидкую консистенцию продуктов (это помогает избежать аспирации) и употреблять пищу часто небольшими порциями.

Ортопедическая помощь
Проблемы со скелетом, связанные со слабыми мышцами при СМА, включают в себя «тугость» суставов с ограниченным диапазоном движений, вывихи бедра, деформацию позвоночника, остеопению, повышенный риск переломов и боли. Слабость мышц, которые обычно стабилизируют суставы в позвоночнике, приводит к развитию кифоза и (или) сколиоза, контрактуры сустава. Большое значение в развитии осложнений имеет осанка, а также положение на кровати или в инвалидной коляске. Люди с СМА также могут извлечь большую пользу из различных форм физиотерапии, трудотерапии, ЛФК с тренером.
Поддержка мобильности
Ортопедические устройства могут использоваться для поддержки тела и для облегчения ходьбы. Например, ортопедические аппараты, такие как AFO (ортезы голеностопного сустава), используются для стабилизации стопы и для облегчения походки, TLSO (грудной пояснично-крестцовый ортез) используется для стабилизации туловища.
Медикаментозное лечение
Nusinersen (Spinraza) – единственный одобренный препараты для лечения мышечной атрофии позвоночника. Он вводится непосредственно в центральную нервную систему с помощью интратекальной инъекции. Препарат был одобрен Европейской комиссией в централизованном порядке в июне 2017 года.
Nusinersen модулирует альтернативный сплайсинг гена SMN2, функционально превращая его в ген SMN1, тем самым повышая уровень белка SMN в ЦНС. Препарат распространяется по ЦНС и периферическим тканям. Основной путь выведения, вероятно, заключается в выделении с мочой нусинерсена и его метаболитов.
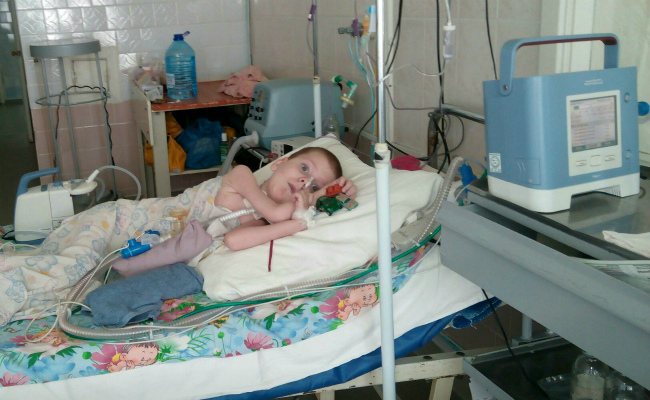
Клиническая картина
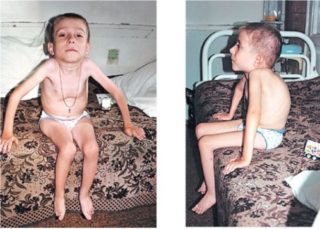
Ребенок со спинальной мышечной атрофией
Симптомы спинальной мышечной атрофии:
- отсутствие рефлексов в ногах и руках;
- задержка в развитии: позднее сидение, стояние, ходьба или вовсе невозможность осуществления подобных действий;
- занятие позы «лягушка» маленькими детьми: в сидячем положении бедра отведены в стороны, а колени согнуты;
- снижение тонуса дыхательных мышц, слабый кашель, ослабленный крик и плач у новорожденного;
- застойный процесс в органах дыхательной системы, задержка бронхиального секрета, которая вызывает дыхательную недостаточность;
- тремор языка;
- нарушение процесса сосания, глотания, кормления.
Выделены 4 формы заболевания, каждая из которых имеет свои симптомы:
- Болезнь Вердинга-Гоффмана (1 тип). Начинает развиваться уже во время пренатального пребывания, первые симптомы возникают в 5-6 месяцев после рождения. Характерные признаки – слабость мышц, снижение рефлексов, выраженное затруднение процесса сосания, дыхания, глотания. Продолжительность жизни – до 1 года в 95% случаев. Летальный исход обусловлен дыхательной недостаточностью.
- Промежуточная амиотрофия Дубовица (2 тип). При заболевании второго типа первые клинические признаки обнаруживают в 3-15 месяцев от рождения. Менее 25% грудничков могут сидеть, но нарушен процесс ходьбы и ползания. Для патологии характерны симптомы паралича, выпадение глубоких сухожильных рефлексов, нарушение глотания. Многие больные уже к 2-3 годам становятся инвалидами и вынуждены находиться в инвалидной коляске. Продолжительность жизни при спинальной мышечной атрофии 2 типа – до 5 лет.
- Спинальная амиотрофия Кугельберг-Веландера (3 тип). Первые симптомы возникают в возрасте 15 месяцев-19 лет и схожи с клинической картиной, присущей 1 типу заболевания. Отличие – медленная прогрессия и лучший прогноз.
- Спинальная амиотрофия 4 типа или взрослая форма. Слабость мышц прогрессирует медленно, в патологический процесс в основном вовлекаются проксимальные мышцы.
У взрослых спинальную амиотрофию разделяют на следующие виды:
- болезнь Кеннеди (амиотрофический боковой склероз);
- болезнь Арана-Дюшенна (хронический полиомиелит взрослых);
- болезнь Шарко-Мари-Тута (моторно-сенсорная нейропатия с поражением дистальных отделов конечностей).
Причина патологии
Возникновение болезни связано с дефицитом или полным отсутствием белка SMN, который участвует в выживаемости двигательных нейронов. Среди факторов риска:
- рождение мертвых детей у ближайших родственников по женской линии в анамнезе;
- смерть новорожденных детей в первый месяц жизни у ближайших родственников;
- наличие дефектного гена у близких родственников;
- диагностируемое заболевание у старших детей.
Первые сведения о спинальной мышечной атрофии появились в конце 19 века в творениях Вердинга-Гоффмана. Им были обнаружены патологические изменения в мышечных волокнах, периферических нервах, спинном мозге у малышей, которые родились с расстройством двигательной функции. Он отметил, что амиотрофия симметрична и развивается в передних рогах и передних корешках.
Механизм СМА
СМА вызывается поломкой в гене SMN1. Ген SMN2 частично компенсирует утрату гена SMN1.
Что происходит?
- Ген SMN1 поврежден и не соответствует норме
- Важные белки не производятся в достаточном количестве
- Двигательные нейроны функционируют неправильно и отмирают
- Импульсы не распознаются
- Мышцы теряют силу и атрофируются
- Движения ограничены, это затрудняет перемещение, дыхание и глотание
Диагностика и помощь специалистов
При выявлении симптомов, которые могут указывать на болезнь, необходимо комплексное обследование для установления точного диагноза.
Диагностика: необходимо получить консультацию невролога, специалиста по нервно-мышечным заболеваниям, который может установить диагноз на основании симптомов. Для подтверждения диагноза требуется проведение ДНК-диагностики и консультация генетика.
Генетическая диагностика: тест ДНК для выявления делеции гена SMN1 и определения количества копий SMN2.
Дополнительные исследования:
- Биохимия крови: креатинкиназа (КФК) — в норме при СМА I, в норме или незначительно повышена при других типах;
- Электронейромиография — показывает снижение нервных импульсов, помогает дифференцировать СМА от других нервно-мышечных болезней. Сенсорная нервная проводимость обычно в норме.
Медицинская помощь
Врачи-специалисты:
Невролог ― ставит диагноз, назначает поддерживающее лечение, ведет постоянное наблюдение за ходом болезни.
Генетик ― ставит диагноз и, в случае необходимости, консультирует семью по вопросам дальнейшего потомства.
Участковый педиатр (терапевт) ― помогает лечить болезни, которыми болеют все.
Пульмонолог или реаниматолог ― помогает выявить и скомпенсировать дыхательные нарушения, решать проблемы с откашливанием, дает консультации по респираторной поддержке.
Ортопед ― оценивает нарушения опорно-двигательного аппарата (контрактуры, деформации), определяет необходимый объем профилактических мер, помогает корректировать эти нарушения.
Нейрохирург ― занимается исправлением сколиоза.
Физический терапевт ― подбирает комплекс упражнений и абилитационных процедур и обучает родителей регулярно делать их дома самостоятельно.
Нутрициолог или диетолог ― помогает подобрать оптимальное питание.
Гастроэнтеролог ― помогает в случае возникновения проблем с желудком и кишечником.
Кардиолог ― наблюдает за работой сердечно-сосудистой системы.
Специалист по паллиативной помощи ― помогает комплексно улучшить качество жизни.
Другие специалисты привлекаются по мере возникновения специфических проблем.
ВАЖНЫЕ АСПЕКТЫ:
Семейно-ориентированный подход ― врач учитывает мнение семьи по всем вопросам, касающимся лечения ребенка, в том числе проведения медицинских вмешательств и их объема, их приемлемости и сроков проведения;
Ориентация на качество жизни пациента ― прежде чем предложить семье применение каких-либо медицинских технологий, необходимо учесть, как это повлияет на качество жизни, потому что важно жить полноценно, а не существовать;
Качественная коммуникация и полноценное информирование пациента и членов семьи обо всех аспектах СМА ― как можно более полная информация о болезни, о том, что будет происходить и с чем придется столкнуться, а также как с этим справляться;
Обучение практическим навыкам ухода и применения медицинского оборудования должно быть неотъемлемой частью медицинской помощи;
Междисциплинарный и мультипрофессиональный подход ― необходима работа междисциплинарной команды (к примеру, невозможно ограничиться наблюдением невролога и получить весь спектр необходимой помощи для полноценной поддержки).
Специфика
Как правило, первые симптомы амиотрофии Кугельберга-Веландера начинают проявляться с 2-х лет, а в более позднем возрасте это заболевание дает о себе знать крайне редко.
Благодаря тому, что мышечная слабость при спинальной амиотрофии III-го типа развивается довольно медленно, способность к самостоятельному передвижению и самообслуживанию у больных, страдающих данной патологией, сохраняется в течение достаточно продолжительного времени.
Предугадать, как именно отразится данный недуг на состоянии ребенка, крайне сложно. Так, некоторые больные становятся глубокими инвалидами еще в подростковом возрасте, а некоторые способны и в 40 лет сохранять хорошо развитые координационные способности.
Осложнения
К наиболее частым неблагоприятным последствиям можно отнести постоянные падения и ассоциированные с ними патологические переломы, что связано с постепенно нарастающей мышечной атрофией мышц нижних конечностей. При бульбоспинальной амиотрофии Кеннеди нередко наблюдаются осложнения со стороны эндокринной и репродуктивной систем – эректильная дисфункция, первичное бесплодие, сахарный диабет.
Более тяжелые состояния встречаются реже, в основном при болезни Верднига-Гоффмана или на поздних стадиях других амиотрофий. Вследствие выраженной атрофии мышц глотки и дыхательной мускулатуры пища попадает в дыхательные пути (аспирация), развивается дыхательная недостаточность. Возможны грубые костно-суставные деформации, утрата способности к ходьбе и самообслуживанию. В единичных случаях обнаруживается рак грудной железы.
Типы сма
| СМА, тип | Генотип | Клиника |
| 0 (нулевой) | Отсутствует ген SMN 11 копия гена SMN2 | Тяжелая форма болезниСмерть внутриутробно или в первый месяц жизни |
| 1 (первый)spinalatrophytype 1Болезнь Верднига-Гоффмана | Делеция или мутация SMN12 копии SMN 2при болезни Верднига | Синдром «вялого» ребенкаТяжелое течениеСмерть в первые 2-3 года жизни (спинальная мышечная атрофия 1 типа сейчас лечится, результаты продолжительности жизни после лечения будут опубликованы в ближайшие 5 лет) |
| 2 (второй)spinal atrophy type 2Болезнь Дубовица | SMN1 после мутации превращается в SMN2SMN 2 присутствует в виде более 3 копий |
Болеют дети от 1 до 2,5 лет
Моторные навыки редуцируютсяВыживаемость при 009sma до 10-14 лет |
| 3 (третий)spinal atrophy type 3Болезнь Кугельберга-Веландера | Копий SMN2 более 3-хМогут быть мутации в основном гене | Появляется после 30 летДолго сохраняется подвижностьНе влияет на продолжительность жизни |
| 4 (четвертый) spinal atrophy type 4Болезнь Кеннеди | Длинный повтор из трех нуклеотидов ЦАГ в гене андрогенного рецептораНаследование сцепленное с X-хромосомой |
Болеют только мужчины
Симптомы стартуют от 30 до 50 лет |
Как видно, симптомы спинальной атрофии Верднига-Гоффмана являются самой тяжелой наследственной формой патологии. При болезни Верднига развивается синдромокомплекс «вялого» ребенка. При 1 типе сма малыш неподвижен, лежит в позе «лягушки», мышечный тонус снижен во всех группах (характерно для spinal atrophy), гипермобильность суставов (типично для болезни Верднига).
Ребенок при спинальной амиотрофии Верднига-Гоффмана не способен приобретать двигательные навыки. Характерно частое поверхностное дыхание, невозможность провести глубокий вдох. При болезни Верднига-Гоффмана малыш не выполняет функциональные мышечные пробы.
https://youtube.com/watch?v=m0HSTbq9WJA
Спинальная миотрофия Верднига Гоффмана, также как и другие типы мышечных спинальных атрофий не только первого типа имеют патогенетическое лечение. Препарат «Спиназа» позволяет улучшить двигательные навыки детей даже с тяжелой формой сма. Курс применения – пожизненный. Применяется препарат в терапии болезни Верднига, Дубовица и других типах амиотрофий.