Журнал «здоровье ребенка» 1 (28) 2011
Содержание:
- Что такое наследственные нервно-мышечные болезни, и как они проявляются?
- Патологическая анатомия
- Мышечная дистрофия лечение
- Диагностика патологии
- Прогноз
- Симптомы заболевания
- Разновидности
- Что представляет собой заболевание
- Основные формы мышечной дистрофии
- Симптомы заболевания
- Наиболее распространенные типы мышечной дистрофии:Duchenne & Becker’s
Что такое наследственные нервно-мышечные болезни, и как они проявляются?
Эти заболевания имеют прогрессирующее течение, и их можно разделить на две различные группы:
- когда первично поражены мышцы (миопатии), в них находятся дефектные ферменты, аномалии строения белка. Такие болезни именуют также миодистрофиями;
- когда первично поражены периферические, двигательные нейроны, которые расположены в передних рогах спинного мозга. Эти заболевания называют спинальными амиотрофиями в случае поражения тел нейронов, и невральными амиотрофиями – если тела нейронов здоровы, но наблюдается нарушение нервно – мышечной проводимости в аксоне, или нерве.
Наконец, в отдельной статье рассмотрена миастения, которая является удивительным заболеванием: и нервы, и мышцы здоровы, но страдает синапс – месть соединения концевой пластинки нерва с мышечным волокном.
Патологическая анатомия
Морфологические изменения при Миопатии характеризуются нарастающей атрофией скелетных мышц, к-рые уменьшаются в объеме и становятся плотными, бурого цвета вследствие разрастания соединительной ткани или, напротив, увеличиваются в объеме за счет жировой клетчатки.
Рис. 1. Микропрепарат мышцы (поперечный срез) при миопатии Дюшенна в стадии частичной сохранности двигательной функции: беспорядочное расположение разнокалиберных мышечных волокон — атрофированных (1), нормального диаметра (2), единичных гипертрофированных (3); дистрофические изменения в части мышечных волокон (4); разрастание соединительной ткани в эндомизии (5); гематоксилин-эозин; X 200.
Рис. 2. Микропрепарат мышцы (продольный срез) при миопатии Дюшенна в стадии обездвиженности: единичные атрофированные мышечные волокна (1) среди фиброзной и жировой ткани; лимфоидно-гистиоцитарная инфильтрация (2); гематоксилин-эозин; X 200.
При различных формах мышечных дистрофий (псевдогипертрофических, плече-лопаточно-лицевой, тазоплечевой, офтальмоплегической, бульбарно-офтальмоплегической) определяются в основном однотипные гистол, изменения (рис. 1): уменьшение количества мышечных волокон в пучках, резкая диффузная разнокалиберность сохранившихся волокон с преобладанием среди них атрофированных, гиалиновая и вакуольная дистрофия в части мышечных волокон, дискоидный и коагуляционный некроз отдельных волокон, расщепление гипертрофированных волокон, разрастание соединительной и жировой ткани в эндо- и перимизии. В нек-рых мышечных волокнах находят саркоплазматические тельца, саркоплазматические массы, кольцевидные миофибриллы. Изредка встречаются регенерирующие волокна с круглыми сочными ядрами и богатой рибонуклеопротеидами саркоплазмой. Мышечные веретена длительное время остаются неизмененными. В части наблюдений выявляются периваскулярные лимфоидно-гистиоцитарные инфильтраты. По мере нарастания двигательных нарушений отмечается постепенное уменьшение количества мышечных волокон и нивелировка их диаметра за счет резкого уменьшения количества и калибра гипертрофированных волокон. Наиболее быстро эти изменения развиваются при миопатии Дюшенна — злокачественном варианте псевдогипертрофической М. Для доброкачественного варианта — миопатии Беккера — характерно сочетание выраженных процессов липоматоза н склероза с резкой гипертрофией части мышечных волокон. С последней особенностью связывают длительную компенсацию двигательных нарушений у таких больных. В далеко зашедшей стадии М. определяются небольшие островки из атрофированных мышечных волокон на фоне резкого склероза и липоматоза эндо- и перимизия (рис. 2), при этом гистологически не представляется возможным дифференцировать М. с нейромышечной атрофией. У пробандов при биопсии мышц находят единичные атрофированные мышечные волокна преимущественно I типа, пролиферацию ядер с переходом их в центр волокна и незначительное увеличение соединительной ткани в эндомизии.
Наиболее ранние ультраструктурные изменения в мышцах при Миопатии характеризуются утолщением и расщеплением Z-линий с последующим разрушением миофибрилл мышечного волокна. Благодаря фазово-контрастной и электронной микроскопии выявлены дефекты в мембранных системах мышечного волокна. Гистоферменто химически отмечается ослабление реципрокных отношений между гликолитическими и окислительными ферментами в мышечных волокнах различного типа.
В центральной и периферической нервной системе изменений не обнаружено. Характерен кардиосклероз.
Мышечная дистрофия лечение
В современной медицине до сих пор отсутствует средство, с помощью которого возможно остановить процесс атрофии мышц. Основные методы, применяющиеся в лечении мышечной дистрофии, направлены на сохранение подвижности различных частей тела больного в течение как можно большего времени. Другими словами, своевременное лечение замедляет атрофию мышц, не устраняя её.
Если имеются подозрения насчёт наличия мышечной дистрофии у ребёнка, следует обратиться к врачу. При осмотре ребёнка и опросе родителей врач может спрогнозировать заболевание у ребёнка (если в семье уже были случаи заболевания). Если у ребёнка отсутствуют родственники с мышечной дистрофией, ему назначается электромиография, позволяющая оценить функцию нервов в мышцах, обнаружить наличие мышечной дистрофии. Биопсия мышечной ткани также используется как метод диагностики мышечной дистрофии.
Лечение мышечных дистрофий основано на замедлении процессов атрофии в мышцах. Для этого применяются: витамин В1, витамин Е, переливания крови, аминокислоты (лейцин, глутаминовая кислота), внутримышечные инъекции АТФ, определённые биологически активные добавки, введение кортикостероидов, никотиновой кислоты. Народная медицина советует использовать поросшие зёрна пшеницы, ржи, траву спорыша, хвоща, женьшень, пчелиное маточное молочко, корневище топинамбура.
В перспективе рассматривается пересадка больному его же стволовых клеток, взятых из костного мозга или же из скелетных мышц. Однако генная инженерия пока не может добиться положительного результата, так как выделенный учёными ген дистрофина не может быть искусственно введён в мышечные клетки, где находится его дефектная копия.
Для лечения мышечной дистрофии используются некоторые виды терапий, позволяющие улучшить качество жизни больного и в некоторых ситуациях её продолжительность:
— Физическая терапия. Направлена на обеспечение максимально возможной подвижности суставов. Позволяет сохранить их гибкость, подвижность;
— Лечебный массаж для поддержания мышечного тонуса и улучшения кровообращения в поражённой области;
— Назначение сосудорасширяющих препаратов. Сочетается с физиотерапией, оксигенотерапией, бальнеотерапией;
— Мобильные устройства. Различные брекеты поддерживают ослабленные мышцы, держат их в растянутом состоянии, сохраняют гибкость мышц, что замедляет прогрессирование контрактуры. Ходунки, трости, коляски помогают больному сохранить мобильность, быть независимым;
— Вспомогательное дыхание (применение специальных устройств, улучшающих поступление кислорода в организм больного во время сна из-за ослабления дыхательных мышц). Для некоторых больных этого недостаточно, поэтому применяются специальные аппараты, нагнетающие кислород в лёгкие;
— Применение ортопедических аппаратов, что укрепляет «висячие» стопы и стабилизирует голеностопные суставы, частоты падений уменьшается;
— Назначение анаболических гормонов. Принимаются данные средства короткими курсами (например, Ретаболил — 1 раз в неделю, курс состоит из 5-6 инъекций) вместе с переливанием крови (по 100 мл);
— При наличии ярко выраженных миотонических симптомов назначается курс Дифенина (0,03-0,05 г 3 раза в сутки, курс применения – около 2,5 недель) для снижения посттетанической активности в мышечной ткани.
Операционное вмешательство для лечения мышечной дистрофии возможно при:
— Наличии контрактур. Операция на сухожилиях ослабляет контрактуры;
— Сколиозе. В данном случае хирургическое лечение применяется для устранения искривления позвоночного столба, затрудняющего дыхание;
— Проблемах с сердцем. Для обеспечения более ритмичного сокращения сердца вводят кардиостимулятор.
Если в семье имеются случаи мышечной дистрофии, необходимо провести медико-генетическую консультацию для того, чтобы выяснить возможное обнаружение заболевания у будущего поколения.
Диагностика патологии
Диагностировать миодистрофию чаще всего удается по результатам опроса родителей. При подозрении на это заболевание проводится физикальное обследование.
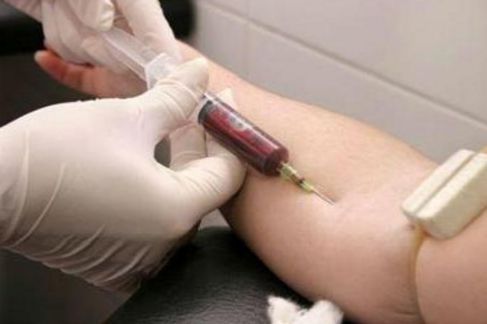 Взятие анализов крови для установления диагноза обязательно
Взятие анализов крови для установления диагноза обязательно
Важной составляющей комплексного обследования для получения полной картины является взятие крови на анализ. По его результатам определяют уровень креатинфосфокиназ
Этот фермент присутствует и в здоровых мышцах, но при миодистрофии его уровень существенно повышается.
Физикальное обследование — это проведение электромиографии. По ее результатам можно определить электрическую активность мышц. Структурные нарушения мышечной ткани определяются путем взятия небольшого ее образца на исследование методом биопсии. По ее результатам у больных миодистрофией не только определяется нарушение структуры, но и повышенное содержание жировых клеток.
Обязательно проводится эхокардиография, которая обеспечивает обнаружение признаков поражения сердечной мышцы. Диагностика обязательно должна быть комплексной, чтобы обнаружить любое поражение.
Прогноз
Прогноз определяется быстротой прогрессирования дистрофических изменений в скелетной мускулатуре, а также возрастом, в к-ром начинается заболевание. При наиболее злокачественной форме миопатии Дюшенна уже в детском возрасте развивается полная обездвиженность больных. Летальный исход может быть обусловлен нарастанием легочной и сердечной недостаточности, гиповентиляционной и гипостатической пневмонией. При поздних формах, начинающихся после 20— 30 лет, течение относительно доброкачественное, больные могут длительное время сохранять трудоспособность. Миотубулярная, немалиновая М., «болезнь центрального стержня», митохондриальные Миопатии обычно отличаются медленным прогрессированием, с возрастом могут приобретать стационарный характер.
Библиография: Гаусманова-Петрусевич И. Мышечные заболевания, пер. с польск., Варшава, 1971; Копьева Т. Н. и Потомская Л. 3. Особенности метаболизма скелетных мышц при прогрессирующей дистрофии формы Дюшенна, Арх. патол. , т. 36, в. 2, с. 35, 1974, библиогр.; Крыжановский Г. Н., Поздняков О. М. и Полгар А. А. Патология синаптического аппарата мышцы, М., 1974; Миопатии, под ред. С. Божинова и др., пер. с болг., София, 1977, библиогр.; Наследственные болезни нервно-мышечной системы, под ред. Л. О. Бадаляна, М., 1974; Adams R. Diseases of muscle, N.Y., 1975; Bethlem J. Muscle pathology, Amsterdam -L., 1970; Dubowitz Y. a. Brooke M. H. Muscle biopsy, a modern approach, L., 1973; Mair W. G. P. a. Tome F. M. S. Atlas of the ultrastructure of diseased human muscle, Edinburgh — L., 1972; New developments in electromyography and clinical neurophysiology, ed. by J. E. Desmedt, v. 1-3, Basel, 1973.
Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Характеристики наиболее распространенных патологий
 Мышечная миопатия Дюшенна. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Мышечная миопатия Дюшенна. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
https://youtube.com/watch?v=zctGZG1JgzY
Миопатия Беккера. Симптомы сравнимы с симптомами М. Д. Дюшенна , однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
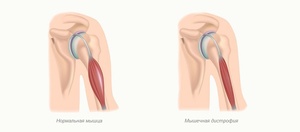 Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Также наблюдается Миотония — аномальное и продолжительное сокращение мышц (мышца расслабляется слишком медленно), особенно выражается в руках, а иногда и на языке. Также могут быть затронуты мышцы лица, шеи и лодыжек. Часто присутствуют сердечные и дыхательные нарушения, которые являются потенциально серьезными. Нередко наблюдаются пищеварительные, гормональные, глазные расстройства, а также бесплодие и раннее облысение.
Миопатия поясничного отдела. Симптомы обычно проявляются в детстве (10 лет) или в раннем взрослом возрасте (около 20 лет). Мышцы плеч и бедер постепенно ослабевают, в то время как мышцы головы, шеи и диафрагмы обычно не затрагиваются. Если некоторые формы сопровождаются дыхательными нарушениями, то при этом типе дистрофии такие аномалии отсутствуют. Сердечные нарушения встречаются редко. Эволюция (развитие заболевания) очень изменчива, в зависимости от формы.
Миопатия Дежерина-Ландузи или плечелопаточная дистрофия. Симптомы обычно появляются в позднем детстве или в зрелом возрасте (от 10 до 40 лет). Как следует из названия, миопатия затрагивает мышцы лица, плеч и рук. Таким образом, больному становится сложно выразить улыбку, произнести некоторые предложения и закрыть глаза. Потеря подвижности происходит примерно в 20% случаев. Заболевание развивается медленно, продолжительность жизни нормальная.
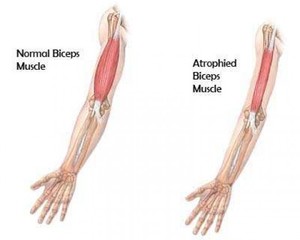 Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Окуло-глоточная миотония. Это заболевание относительно распространено в Квебеке. Симптомы обычно появляются около 40 или 50 лет. Первые признаки болезни проявляются опустившимися веками, за которыми следуют слабость мышц глаз, лица и горла (глотки), вызывая трудности с глотанием пищи. Прогрессирование заболевания происходит медленно.
https://youtube.com/watch?v=Rnjsh0bmHXA
Разновидности
Клиницисты выделяют несколько наиболее распространённых форм недуга.
Мышечная дистрофия Дюшена. Данную разновидность также именуют в медицинской литературе псевдогипертрофической. Обычно мышечная дистрофия Дюшена начинает прогрессировать уже в детском возрасте. Примечателен тот факт, что этому недугу более подвержены маленькие мальчики, нежели девочки.
Симптомы мышечной дистрофии Дюшена проявляются у малышей уже в возрасте от 2 до 5 лет. Изначально поражаются мышцы ног, тазового пояса. Но постепенно патологический процесс «перебирается» на мышцы верхней части туловища. Позже вовлекаются и остальные мышечные структуры. Примечателен тот факт, что недуг стремительно прогрессирует, и уже в среднем к 12–15 годам больной полностью утрачивает способность свободно передвигаться. Прогноз этого типа дистрофии не является благоприятным – многие люди погибают, даже не достигнув 20-летнего возраста.
Прогрессирующая мышечная дистрофия Дюшена
Болезнь Штейнерта. Эта разновидность патологии является характерной для взрослых людей из возрастной категории от 20 до 40 лет. Редки случаи, когда патология проявляет себя уже в младенческом возрасте. Ограничений, касательно половой принадлежности, дистрофия не имеет. Характеризуется медленным прогрессированием.
Дистрофия этого вида имеет свою характерную черту – патологический процесс затрагивает не только мышцы скелета, но также и структуры жизненно важных органов. У больного возможно появление слабости мышц лица. Стоит отметить, что поражение других групп мышц также не исключено. Характерно медленное расслабление волокон после их предварительного сокращения.
Прогрессирующая мышечная дистрофия Беккера. Эта разновидность патологии является малораспространенной. Патологический процесс прогрессирует довольно медленно. Прогрессирующую мышечную дистрофию Беккера обычно диагностируют у людей с низким ростом. Прогноз недуга благоприятный. На протяжении многих лет люди с таким диагнозом сохраняют свою работоспособность, и их состояние остаётся удовлетворительным. Инвалидизации способствуют травмы различной степени тяжести, а также сопутствующие болезни.
Юношеская мышечная дистрофия Эрба-Рота. Период проявления её симптомов – от 10 до 20 лет. Прогрессирует медленно. На начальных стадиях развития отмечается атрофия волокон рук и плеч, позже – ног и таза. Во время ходьбы можно отметить изменение осанки человека – грудная клетка отодвигается немного назад, в то время как живот выпячивается вперёд. Больной идёт и переваливается.
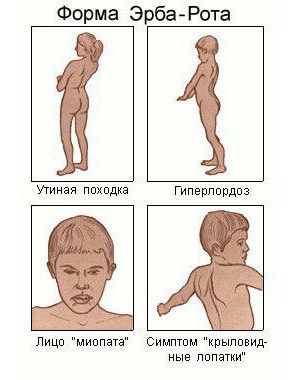
Мышечная дистрофия Эрба-Рота
Мышечная дистрофия Ландузи-Дежерина. Симптомы недуга проявляются в период от 6 до 52 лет. Чаше всего признаки дистрофии Ландузи-Дежерина выявляют в период с 10 до 15 лет. При этом недуге в первую очередь страдают мышцы лица. Но постепенно при дистрофии Ландузи-Дежерина патологический процесс охватывает также мышечные структуры конечностей и туловища.
Первым симптомом развития болезни является неполное смыкание век. Постепенно перестают полностью смыкаться губы, что обуславливает нарушение дикции. Патология Ландузи-Дежерина у больных протекает достаточно медленно. У пациента длительное время сохраняется способность двигаться, поэтому он может вести нормальный образ жизни. В среднем через 20–25 лет возможно атрофирование мышц тазового пояса, что и становится причиной инвалидизации. В целом можно сказать, что дистрофия Ландузи-Дежерина протекает благоприятно.
Что представляет собой заболевание
Эндокринная офтальмопатия (орбитопатия) — тяжёлая патология, при которой помощь пациенту оказывают эндокринологи и офтальмологи. Болезнь имеет аутоиммунную природу, чаще всего возникает на фоне нарушений со стороны щитовидной железы и представляет собой поражение подкожной клетчатки и мышц, окружающих глазные яблоки. Впервые патология была описана Грейвсом ещё в позапрошлом веке, поэтому её часто именуют офтальмопатией Грейвса. Ещё недавно болезнь считалась симптомом токсического зоба, сопровождающегося повышенной выработкой тиреотропных гормонов и приводящего к тиреотоксикозу. Сейчас эндокринная орбитопатия выделена в самостоятельное заболевание.
По статистике, женщины болеют намного чаще мужчин, причём поражаются люди сорокалетнего или шестидесятилетнего возраста. В медицинской литературе описаны случаи заболевания у детей. Лёгкие формы болезни чаще бывают у молодых людей, для пожилого возраста характерно развитие тяжёлых форм офтальмопатии.

Орбитопатия Грейвса проявляется характерными признаками и чаще всего возникает на фоне патологий щитовидной железы
В 80–90% случаев болезнь протекает на фоне гормональных дисфункций со стороны щитовидной железы (гипотиреоз, аутоиммунный тиреоидит, тиреотоксикоз). При этом проявления со стороны глаз могут развиваться сразу с клиническими симптомами поражения железы, а могут предшествовать им или даже появляться в отдалённом времени (через 5–10 лет после лечения щитовидки). В 6–25% случаев офтальмопатия может возникать на фоне эутиреоза (состояния, при котором щитовидная железа работает правильно и уровень тиреоидных гормонов соответствует норме).
Основные формы мышечной дистрофии
Дистрофия Дюшенна
Преимущественно болеют ею мальчики в возрасте от 2 до 5 лет. В начале поражаются мышцы нижних конечностей и таза, дальше болезнь охватывает и остальные мышцы. Утолщаются икроножные мышцы, к 12 годам ребенок утрачивает способность к движению. К 20 годам возможен летальный исход.
Болезнь Штейнерта
Возникает в период от 20 до 40 лет, иногда бывает и в младенчестве.
Основной признак — после сокращения наблюдается замедленное расслабление напряженной мышцы. Отмечается слабость мышц лица, возможно поражение и других групп мышц. Нередко страдают мышцы внутренних органов, в том числе сердечная мышца, и скелетные мышцы. Характерно медленное прогрессирование.
Дистрофия Беккера
Зафиксирована у низкорослых людей. Редко встречается, медленно развивается. При сопутствующих заболеваниях, а также травмах начинает прогрессировать.
Дистрофия Эрба-Рота
Эта юношеская форма заболевания иногда проявляется в возрасте от 10 до 20 лет.
Основное отличие в очередности поражений. Заболевание начинается с мышц плечевого пояса и рук, дальше страдают нижние конечности и мышцы таза. У больного меняется походка: выпячивается живот, отодвигается назад грудная клетка.
Дистрофия Ландузи-Дежерина
Характерно поражение мышц лица, плечевого пояса, рук, позже ног и таза. Чаще всего болезнь проявляется в 10-15 лет. Хотя иногда встречаются пациенты от 6 лет до 52 .
На начальном этапе плохо смыкаются веки, наблюдаются некоторые проблемы с дикцией из-за поражения губных мышц. Болезнь развивается медленно. Через 15-25 лет дистрофия поражает мышцы таза и ног.
Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Характеристики наиболее распространенных патологий
Мышечная миопатия Дюшенна. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
https://youtube.com/watch?v=zctGZG1JgzY
Миопатия Беккера. Симптомы сравнимы с симптомами М. Д. Дюшенна , однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Также наблюдается Миотония — аномальное и продолжительное сокращение мышц (мышца расслабляется слишком медленно), особенно выражается в руках, а иногда и на языке. Также могут быть затронуты мышцы лица, шеи и лодыжек. Часто присутствуют сердечные и дыхательные нарушения, которые являются потенциально серьезными. Нередко наблюдаются пищеварительные, гормональные, глазные расстройства, а также бесплодие и раннее облысение.
Миопатия поясничного отдела. Симптомы обычно проявляются в детстве (10 лет) или в раннем взрослом возрасте (около 20 лет). Мышцы плеч и бедер постепенно ослабевают, в то время как мышцы головы, шеи и диафрагмы обычно не затрагиваются. Если некоторые формы сопровождаются дыхательными нарушениями, то при этом типе дистрофии такие аномалии отсутствуют. Сердечные нарушения встречаются редко. Эволюция (развитие заболевания) очень изменчива, в зависимости от формы.
Миопатия Дежерина-Ландузи или плечелопаточная дистрофия. Симптомы обычно появляются в позднем детстве или в зрелом возрасте (от 10 до 40 лет). Как следует из названия, миопатия затрагивает мышцы лица, плеч и рук. Таким образом, больному становится сложно выразить улыбку, произнести некоторые предложения и закрыть глаза. Потеря подвижности происходит примерно в 20% случаев. Заболевание развивается медленно, продолжительность жизни нормальная.
Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Окуло-глоточная миотония. Это заболевание относительно распространено в Квебеке. Симптомы обычно появляются около 40 или 50 лет. Первые признаки болезни проявляются опустившимися веками, за которыми следуют слабость мышц глаз, лица и горла (глотки), вызывая трудности с глотанием пищи. Прогрессирование заболевания происходит медленно.
https://youtube.com/watch?v=Rnjsh0bmHXA
Наиболее распространенные типы мышечной дистрофии:Duchenne & Becker’s
Мышечная дистрофия Дюшенна:
- Эта форма мышечной дистрофии поражает мужчин с рождения. Обычно он начинает вызывать симптомы в раннем детстве, обычно в возрасте до 5 лет. По данным группы защитников и веб-сайта для родительского проекта «Мышечная дистрофия» (PPMD), Дюшенн является наиболее распространенным фатальным генетическим заболеванием, диагностируемым в детстве. Это влияет примерно на 1 на каждые 3500 живорожденных мужчин.
- Ежегодно во всем мире диагностируется около 20 000 новых случаев. Дюшенн поражает только новорожденных мужчин, поскольку ген Дюшенна находится в Х-хромосоме. Самки могут быть носителями дистрофии Дюшенна, передавая их своим потомкам, но на самом деле сами не страдают от этого расстройства.
- Мутация в гене дистрофина, который кодирует белок, называемый дистрофином, вызывает Дюшенна. Дистрофин помогает поддерживать здоровые структурные элементы мышечной ткани и клеточных мембран. Без дистрофина происходит прогрессирующая мышечная слабость и, в конечном итоге, смерть.
- Симптомы мышечной дистрофии обычно появляются в младенчестве или у детей до 5 лет. Некоторые из них не имеют диагноза до появления первых симптомов. Тем не менее, все большее число лабораторных тестов используются для выявления тех, кто может иметь заболевание при рождении.
Мышечная дистрофия Беккера:
- Подобная мутация гена Дюшенна вызывает у Беккера, но она менее серьезна. Те, у кого Дюшенн, не производят дистрофина, а те, у кого Беккер. Тем не менее, дистрофин работает не так, как обычно, и его уровни ниже, чем у здоровых людей без каких-либо расстройств.
- Мышечная дистрофия Беккера обычно начинается позже, чем у Дюшенна, когда ему около 12 лет. Хотя он также прогрессирует, он обычно менее серьезен, чем Дюшенн, и поэтому вызывает менее серьезные симптомы. Тем не менее, люди с дистрофией Беккера все еще обычно имеют слабость, болезни сердца, проблемы с искривлением позвоночника, усталость, проблемы с мышлением и затрудненное дыхание.